Chapter 20 Renal tubular acidosis
20.1 Classification of RTA
type II (pRTA) = impaired reabsorption of HCO3
type I (dRTA) = inability to secrete H+:
- complete = systemic acidosis
- incomplete = no systemic acidosis
- complete = systemic acidosis
type IV = hypoaldosteronism (+/- impaired ammoniagenesis from hyperK)
type III (mixed) = CA inhibition
RTA in CKD:
- HCMA when GFR < 30
- WGMA when GFR < 15
20.1.1 Pathogenesis of hypokalaemic RTA
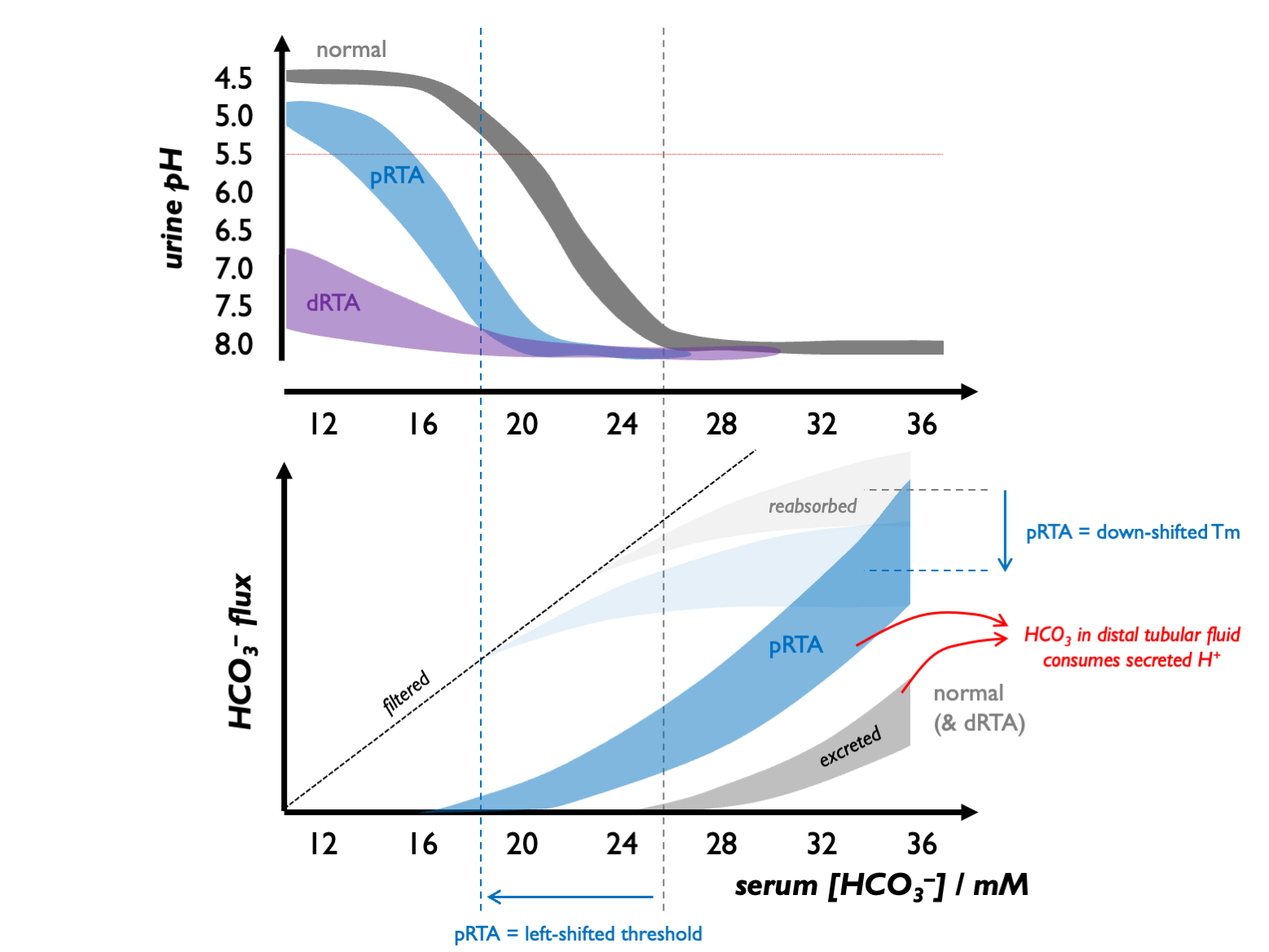
Normally, the tubular threshold for HCO3 is around 24 - 26 mM, so that plasma [HCO3] is maintained in that range.
In dRTA, there is no change in the tubular threshold for HCO3 but due to a failure of distal acidification, urine pH never falls even in the face of systemic acidosis.
In pRTA, there is a reduction in the apparent Tm (and hence tubular threshold) for HCO3. As a consequence, in mild systemic acidosis, there is renal bicarbonate wasting, maintaining an alkaline urine. However, as the acidosis becomes more profound and [HCO3] drops below the tubular threshold, there is no tubular HCO3 loss and - because distal acidification remains intact - the urine can be acidified.
(These mechanisms were established by Edelman and colleagues in children with RTA in the late 1960s.)
 Therefore:
Therefore:
in dRTA: urine pH is never low (always well above 5.5); a negative acid balance can never be achieved - hence the severe skeletal phenotype
in pRTA: urine pH may be alkaline or acid - depending on how profound the systemic acidosis is; negative acid balance can be achieved, avoiding a very severe skeletal phenotype
Furthermore:
in dRTA: relatively modest HCO3 supplementation is required in dRTA (in the order of 1 mmol per kg per day to regenerate that lost to the buffering of non-volatile acid)
in pRTA: absolutely massive HCO3 supplementation - above 10 mmol per kg per day - would be required to drive systemic [HCO3] into the normal range; this might end up being counter-productive if the associated bicarbonaturia drives excessive K+ and Na+ loss
20.1.2 Pathogenesis of hyperkalaemic RTA
Two mechanisms account for type IV RTA:
reduced mineralocorticoid activity in the distal nephron, meaning that there is less electrogenic Na+ reabsorption (through ENaC) and a diminished electrical force driving H+ and K+ secretion;
hyperkalaemia per se impairs ammoniagenesis in the PCT
20.1.3 Associations in dRTA
Classically:
- CaP stones (alkaline urine, hypocitraturia, hypercalciuria)
- nephrocalcinosis (near-universal in inherited forms)
- mild-moderate bone disease (rickets / osteomalacia / osteopenia)
Also often present but under-appreciated:
- urinary concentrating deficit (from nephrocalcinosis and hypoK) - therefore vulnerable to dehydration
- CKD from nephrocalcinosis
20.1.4 Associations in pRTA
- severe rickets / osteomalacia (due to phosphate wasting)
- Fanconi syndrome
…but NOT stones / nephrocalcinosis
20.1.5 Investigation of suspected RTA
UAG & FENaHCO3
urine pH (never < 5.5 in dRTA)
in suspected pRTA
- tubular reabsorption of phosphate (TRP = 100 - FEPO4) < 85 %
- glycosuria, aminoaciduria, LMW proteinuria
in suspected dRTA
- furosemide-fludrocortisone (FF) test (failure to achieve pH < 5.3 , 3 – 4 hrs after 40 – 80 mg / 1 mg)
- UCa > 4 mg / kg / day (or spot UCa/Cr > 0.2) in type I RTA
- USS / KUB (medullary nephrocalcinosis in type I RTA)
- urinary citrate (low in type I RTA; high in type II / IV RTA)
kidney biopsy (to detect subclinical TIN)
Test urinary RBP (retinol binding protein) as the most sensitive marker of proximal renal tubular dysfunction - good in sarcoid, Sjogren’s etc. Good for diagnosis and for tracking disease activity.
USS more sensitive than CT for nephrocalcinosis in the context of hypoparathyroidism, but CT more specific (Boyce JCEM 2013). Plain films very insensitive. Therefore prefer USS for screening (but consider CT for verification).
20.1.6 Causes
| PRTA (ISOLATED) | inherited | inherited |
| acquired | topiramate | |
| PRTA (WITH FANCONI) | inherited | NBCe1A (AR with ocular abnormalities) |
| Wilson’s | ||
| cystinosis | ||
| fructose intolerance | ||
| Dent disease | ||
| mitochondrial cytopathies | ||
| myeloma | ||
| LCDD | ||
| LCFS is κ in 96 %, | ||
| amyloid | ||
| Sjogren’s | ||
| other TIN | ||
| allograft rejection | ||
| tenofovir | ||
| lamivudine | ||
| aminoglycosides | ||
| outdated tetracyclines | ||
| cisplatin | ||
| valproate | ||
| lenalinomide | ||
| Pb | ||
| Hg | ||
| Cd | ||
| aristolochic acid | ||
| DRTA | inherited | AEI (AR) |
| H-ATPase B1 (AR with SNHL) | ||
| H-ATPase A4 (AR) | ||
| chronic pyelonephritis | ||
| chronic TIN | ||
| obstructive uropathy | ||
| sickle cell | ||
| allograft rejection | ||
| hypergammaglobulinaemia | ||
| SLE | ||
| Sjogren’s | ||
| chronic active hepatitis | ||
| PBC | ||
| Li+ | ||
| amphoterocin | ||
| toluene | ||
| hyperPTH | ||
| idiopathic hypercalciuria | ||
| MSK | ||
| TYPE IV | low renin | DM |
| NSAIDs | ||
| CNIs | ||
| β– | ||
| Addison’s | ||
| CAH | ||
| ACEi / ARB | ||
| heparin | ||
| ketoconazole | ||
| TIN | ||
| sprionolactone / amiloride | ||
| trimethoprim | ||
| TYPE III | inherited | CAII (AR with osteopetrosis / cerebral calcification) |
| topiramate (CA inhibition) |
Inherited dRTA caused by mutations in A-IC proteins (see here & here):
- apical H+-ATPase subunits (ATP6V1B1, ATP6V0A4) > AR
- basolateral HCO3-Cl exchanger (AE1 aka SLC4A1) > AD/AR
- transcription factor regulating H+-ATPase expressuib (FOXI1) > AR
- unknown (WDR72) > AR
All of the mutations that can affect H+-ATPase function (i.e. including FOXI1) may have SNHL. SNHL nearly universal in ATP6VB1; around 50% in ATP6V04. Some AE1 mutations associated with haemolytic anaemia (not all because of alternative splicing in red cell and kidney isoforms).
AR forms almost always presents with failure-to-thrive in first year of life. AD SLC4A1 mutations and AR WDR72 mutations are milder phenotype: typically present in primary school and often by screening in family members.
AD SLC4A1 disease (the commonest form) typically diagnosed in adolescence / adulthood.
Similarly, idRTA (incomplete) usually in adults: suspect if nephrocalcinosis / stones and borderline HCO3 / hypocitraturia. K > 3.8 mM is strongly predictive against this diagnosis.
(Intriguingly, there is a case report of gain-of-function H+-ATPase mutations causing a renal tubular alkalosis!)
Acquired causes of dRTA:
- pSS
- other autoimmune (SLE, PBC, AIH, thyroiditis)
- MSK
- nephrocalcinosis (e.g. FHCCN)
- drugs
Sjogrens classically causes a dRTA (but can also cause pRTA if interstitial nephritis). Autoimmune dRTA is Sjogrens in 80 - 90%. Subclinical tubular involvment in 30% Sjorens. Often associated with hypocitruria and low-grade features of Fanconi syndrome. Treat CIN with MMF or pred / RTX. Treat dRTA with K citrate. Autoantibodies to H+-ATPase or bicarb exchanger.
Mitochondrial cytopathy: raised CK, abnormal number of microchondria (EM).
There is a complex relationship between vitamin D deficiency and RTA. There are several case reports / case series showing that these co-exist. Elements of this relationship include:
secondary hyperPTH in vitD deficiency will drive proximal tubular phosphate and bicarbonate wasting
severe vitD deficiency can be associated with Fanconi syndrome and vitD replacement has in some cases corrected the Fanconi syndrome (i.e. consistent with a causal link)
co-existent vitD deficiency and dRTA can potentiate the effects of both on bone causing severe osteomalacia
20.2 Fanconi syndrome
In adults, usually acquired:
- tenfovir and other drugs (antivirals, fumaderm for Psoriasis)
- heavy metals
- LCCD
- post-Tx
Check for:
- LMWH proteinuria (e.g. retinol binding protein; high uPCR with a negative dip)
- uricosuria
- phosphaturia
- renal glycosuria
LMW proteinuria is the most sensitive test (because megalin is the most energy-sensitive process and therefore most vulnerable to becoming disrupted). Glycosuria appears last (least energy-sensitive process).
Interpret tubular proteinuria by plotting uRBP vs. uACR (as per Norden et al., KI 2000).
Inherited pRTA is very rare; inherited dRTA less so. Commonest cause of inherited Fanconi syndrome is cystinosis.
20.2.1 XLD hypophosphataemic rickets
Commonest cause of heritable rickets. PHEX mutations result in excessive production of FGF23 (phosphatonin). Treat with burosumab.
20.2.2 CLCN5 mutations (Dent disease spectrum)
CLCN5 encodes chloride channel involved in endosomal acidification – therefore impaired endocytosis by megalin / cubulin. Defective endocytosis of vitD and PTH lead to calciuria.
In males, get classic XLR disorder:
- proteinuria (may be nephrotic range but no nephrotic syndrome)
- hypercalciuria / nephrocalcinosis / stones / osteomalacia
- hypoK
- unexplained CKD
- FHx of kidney failure in male relatives on maternal side
Proteinuria often includes a lot of albuminuria (presumably reflecting impaired tubular reabsorption of filtered albumin) but uRBP disproportionately high.
(Therefore historically termed XLR nephrolithiasis, XLR hypophosphatamic rickets, idiopathic LMW proteinuria, incomplete Fanconi syndrome: now all recognised as Dent spectrum.)
Diagnose by:
- look for LMWP first (if negative then Dent disease excluded)
- then look for complete Fanconi syndrome (if present than alternative cause more likely)
- then genetic testing (NOT biopsy)
In carrier females (usually unaffected or mild):
- LMW proteinuria
- hypercalciuria
- rarely NVH, stones, nephrocalcinosis, CKD
20.3 Management of adult dRTA
Alkali supplements for RTA (see ESPN clinical practice points, NDT 2021):
- target normal HCO3, Cl, K and a urine Ca-excretion within normal range
- usually 2 - 3 mEq/kg/day alkali in adults
- prefer K-citrate for the reasons here, prefer frequent dosing
- …and sometimes need additional K supplements
- vegan diet (or as vegan as possible)
- DEXA every 2 - 3 yrs in adults
- surveillance USS 1 - 2 yrs (or CTKUB) to detect stones early